VCF parsing#
Introduction#
fastdfe provides parser utilities that enable convenient parsing of frequency spectra from VCF files. By default, Parser looks at the AA tag in the VCF file’s info field to retrieve the correct polarization. Sites for which this tag is not well-defined are by default included (see skip_non_polarized). Note that non-polarized frequency spectra provide little information on the distribution of beneficial mutations, however.
We might also want to stratify the SFS by some site properties, such as site-degeneracy. This is done by passing stratifications to the parser. In this example, we will stratify the SFS by 0-fold and 4-fold degenerate sites using a VCF file for Betula spp.
See also
VCF-to-SFS parsing, annotation, and filtration are now provided by the standalone sfsutils package.
library(fastdfe)
fd <- load_fastdfe()
url <- "https://github.com/Sendrowski/fastDFE/blob/dev/"
# instantiate parser
p <- fd$Parser(
n = 8,
vcf = paste0(url, "resources/genome/betula/biallelic.polarized.subset.50000.vcf.gz?raw=true"),
fasta = paste0(url, "resources/genome/betula/genome.subset.1000.fasta.gz?raw=true"),
gff = paste0(url, "resources/genome/betula/genome.gff.gz?raw=true"),
annotations = c(
fd$DegeneracyAnnotation()
),
stratifications = c(fd$DegeneracyStratification())
)
# parse SFS
spectra <- fd$Parser$parse(p)
INFO:Parser: Using stratification: [neutral, selected].
INFO:Parser: Loading VCF file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/70975cbc50c2.biallelic.polarized.subset.50000.vcf.gz
INFO:Parser: Loading GFF file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/2d03158a125f.genome.gff.gz
INFO:sfsutils: Unzipping /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/2d03158a125f.genome.gff.gz to /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/tmp3tgydv_k.gff
INFO:Parser: Loading FASTA file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/eb1f2b2b0185.genome.subset.1000.fasta.gz
INFO:sfsutils: Unzipping /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/eb1f2b2b0185.genome.subset.1000.fasta.gz to /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/tmpxt1zle5k.fasta
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/70975cbc50c2.biallelic.polarized.subset.50000.vcf.gz
Parser>Counting sites: 50000it [00:00, 55125.27it/s]
Parser>Processing sites: 100%|██████████| 50000/50000 [00:22<00:00, 2258.77it/s]
INFO:PolyAllelicFiltration: Filtered out 0 sites.
INFO:DegeneracyStratification: Number of sites with valid type: 13771
INFO:DegeneracyAnnotation: Annotated 19002 sites.
INFO:Parser: Skipped 1669 sites without ancestral allele information.
INFO:Parser: Included 13771 out of 50000 sites in total from the input.
# visualize SFS
p <- fd$Spectra$plot(spectra)
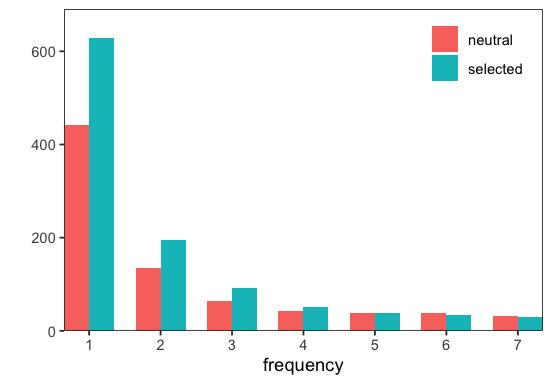
fastdfe relies here on VCF info tags to determine the degeneracy of a site but this behavior can be customized (cf. DegeneracyStratification).
Stratifications#
We can use also several stratifications in tandem by specifying a list of stratifications. In this example, we will stratify the SFS by synonymy as well as base transitions type. The resulting spectra can be fed directly into fastdfe’s inference routines. See the sfsutils stratifications reference for a complete list of available stratifications.
# instantiate parser
p <- fd$Parser(
n = 8,
vcf = paste0(url, "resources/genome/betula/biallelic.polarized.subset.50000.vcf.gz?raw=true"),
fasta = paste0(url, "resources/genome/betula/genome.subset.1000.fasta.gz?raw=true"),
gff = paste0(url, "resources/genome/betula/genome.gff.gz?raw=true"),
annotations = c(
fd$DegeneracyAnnotation()
),
stratifications = c(
fd$DegeneracyStratification(),
fd$AncestralBaseStratification()
)
)
# parse SFS
spectra <- fd$Parser$parse(p)
INFO:Parser: Using stratification: [neutral, selected].[A, C, G, T].
INFO:Parser: Loading VCF file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/70975cbc50c2.biallelic.polarized.subset.50000.vcf.gz
INFO:Parser: Loading GFF file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/2d03158a125f.genome.gff.gz
INFO:sfsutils: Unzipping /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/2d03158a125f.genome.gff.gz to /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/tmpks05jxv0.gff
INFO:Parser: Loading FASTA file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/eb1f2b2b0185.genome.subset.1000.fasta.gz
INFO:sfsutils: Unzipping /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/eb1f2b2b0185.genome.subset.1000.fasta.gz to /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/tmpas8sg5lx.fasta
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/70975cbc50c2.biallelic.polarized.subset.50000.vcf.gz
Parser>Counting sites: 50000it [00:00, 56384.92it/s]
Parser>Processing sites: 100%|██████████| 50000/50000 [00:22<00:00, 2228.69it/s]
INFO:PolyAllelicFiltration: Filtered out 0 sites.
INFO:DegeneracyStratification: Number of sites with valid type: 13771
INFO:AncestralBaseStratification: Number of sites with valid type: 13771
INFO:DegeneracyAnnotation: Annotated 19002 sites.
INFO:Parser: Skipped 1669 sites without ancestral allele information.
INFO:Parser: Included 13771 out of 50000 sites in total from the input.
# visualize SFS
p <- fd$Spectra$plot(spectra)
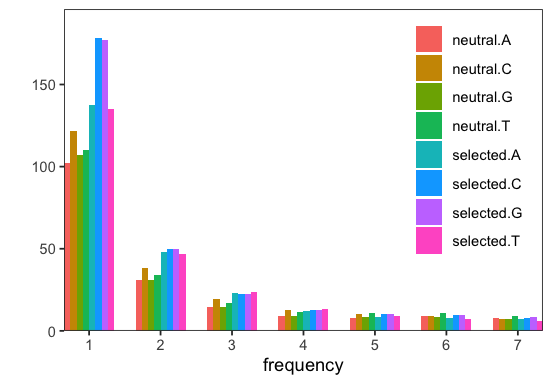
Note that fastdfe requires the ancestral state of sites to be determined. The Parser achieves this by examining the AA field, although this behavior can be customized.
Annotations#
fastdfe provides a number of annotations accessible directly during the parsing process. To annotate a VCF file directly, consider using the Annotator class.
Degeneracy Annotation#
DegeneracyAnnotation annotates the SFS by the degeneracy of the site. This annotation requires information from a FASTA and GFF file and is useful for stratifying the SFS by 0-fold and 4-fold degenerate sites which is what we often want to do when inferring the DFE (see DegeneracyStratification).
# example for degeneracy annotation
ann <- fd$Annotator(
vcf = paste0(url, "resources/genome/betula/biallelic.subset.10000.vcf.gz?raw=true"),
fasta = paste0(url, "resources/genome/betula/genome.subset.1000.fasta.gz?raw=true"),
gff = paste0(url, "resources/genome/betula/genome.gff.gz?raw=true"),
annotations = c(fd$DegeneracyAnnotation()),
output = "genome.deg.vcf.gz"
)
fd$Annotator$annotate(ann)
INFO:Annotator: Start annotating
INFO:Annotator: Loading GFF file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/2d03158a125f.genome.gff.gz
INFO:sfsutils: Unzipping /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/2d03158a125f.genome.gff.gz to /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/tmpe6mvao28.gff
INFO:Annotator: Loading FASTA file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/eb1f2b2b0185.genome.subset.1000.fasta.gz
INFO:sfsutils: Unzipping /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/eb1f2b2b0185.genome.subset.1000.fasta.gz to /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/tmpqcdm4viz.fasta
INFO:Annotator: Loading VCF file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/7a41c74709e8.biallelic.subset.10000.vcf.gz
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/7a41c74709e8.biallelic.subset.10000.vcf.gz
Annotator>Counting sites: 10000it [00:00, 56996.96it/s]
Annotator>Processing sites: 100%|██████████| 10000/10000 [00:02<00:00, 4260.28it/s]
INFO:DegeneracyAnnotation: Annotated 3566 sites.
Ancestral Allele Annotation#
Currently, two ancestral allele annotations are available: MaximumParsimonyAncestralAnnotation and MaximumLikelihoodAncestralAnnotation. The former is straightforward but susceptible to errors, and only appropriate if no outgroup information is available. Alternatively, if outgroups are missing, DFE inference can also be performed on folded spectra, but please note that this will yield less precise estimates. Ideally, we would like to use MaximumLikelihoodAncestralAnnotation, which is more sophisticated and requires one or several outgroup to be specified. Its underlying model is very similar to EST-SFS.
# example for ancestral allele annotation with outgroups
ann <- fd$Annotator(
vcf = paste0(url, "resources/genome/betula/all.with_outgroups.subset.10000.vcf.gz?raw=true"),
annotations = c(fd$MaximumLikelihoodAncestralAnnotation(
outgroups = list("ERR2103730"),
n_ingroups = 10
)),
output = "genome.aa.vcf.gz"
)
fd$Annotator$annotate(ann)
INFO:Annotator: Start annotating
INFO:Annotator: Loading VCF file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/a3b0e449a317.all.with_outgroups.subset.10000.vcf.gz
INFO:Annotator: Loading VCF file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/a3b0e449a317.all.with_outgroups.subset.10000.vcf.gz
INFO:MaximumLikelihoodAncestralAnnotation: Subsampling 10 ingroup haplotypes probabilistically from 378 individuals in total.
INFO:MaximumLikelihoodAncestralAnnotation: Using 1 outgroup samples (ERR2103730).
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/a3b0e449a317.all.with_outgroups.subset.10000.vcf.gz
Annotator>Counting sites: 10000it [00:00, 149794.43it/s]
MaximumLikelihoodAncestralAnnotation>Parsing sites: 100%|██████████| 10000/10000 [00:07<00:00, 1405.17it/s]
INFO:MaximumLikelihoodAncestralAnnotation: Included 6689 sites for the inference (1027 polymorphic, 5662 monomorphic).
WARNING:MaximumLikelihoodAncestralAnnotation: The number of monomorphic sites is unusually low. Please note that including monomorphic sites is necessary to obtain realistic branch rate estimates.
WARNING:MaximumLikelihoodAncestralAnnotation: If your dataset does not contain any monomorphic sites, consider using the `n_target_sites` argument.
MaximumLikelihoodAncestralAnnotation>Optimizing rates: 100%|██████████| 10/10 [00:00<00:00, 11.40it/s]
Annotator>Processing sites: 100%|██████████| 10000/10000 [00:02<00:00, 3492.33it/s]
INFO:MaximumLikelihoodAncestralAnnotation: Annotated 10000 sites.
INFO:MaximumLikelihoodAncestralAnnotation: There were 195 mismatches between the most likely ancestral allele and the ad-hoc ancestral allele annotation.
Filtrations#
fastdfe also offers a number of filtrations which can be accessed immediately while parsing. Alternatively, to filter a VCF file directly, use the Filterer class. Some useful filtrations include DeviantOutgroupFiltration, CodingSequenceFiltration, and BiasedGCConversionFiltration. For a complete list of available filtrations, refer to the API reference.
# example for filtration
f <- fd$Filterer(
vcf = paste0(url, "resources/genome/betula/biallelic.subset.10000.vcf.gz?raw=true"),
filtrations = c(fd$BiasedGCConversionFiltration()),
output = "genome.gc.vcf.gz"
)
fd$Filterer$filter(f)
INFO:Filterer: Start filtering
INFO:Filterer: Loading VCF file
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/7a41c74709e8.biallelic.subset.10000.vcf.gz
INFO:FileHandler: Using cached file at /var/folders/n4/m5q2jgw91zv0tp1c4j9bh48m0000gn/T/7a41c74709e8.biallelic.subset.10000.vcf.gz
Filterer>Counting sites: 10000it [00:00, 54955.01it/s]
Filterer>Processing sites: 100%|██████████| 10000/10000 [00:00<00:00, 10916.31it/s]
INFO:BiasedGCConversionFiltration: Filtered out 7903 sites.
INFO:Filterer: Filtered out 7903 of 10000 sites in total.
Manipulating spectra#
For the full set of operations on the resulting Spectrum and Spectra objects (folding, polarising, resampling, plotting, and serialisation), see the Manipulating the SFS guide in the sfsutils documentation.